There are many approaches of computational chemistry which are popular in molecular modeling in biological sciences:
There is a justified interest in Density Functional Approaches among chemists.
While they have been a domain of physicists for a long time, these methods have now
found their way into mainstream chamistry. The traditional ab initio
approaches, which are the workhorse of Quantum Chemistry offer a prescription
for calculated truth. In principle, one can calculate chemical properties
with any desired accuracy. The methods are known and proven to work.
The only problem is that for systems larger than hydrogen or lighter H![]() X molecules,
the calculations involved in obtaining accurate results are frequently impractical.
We know how to compute it, but we do not have computational power to do it (probably
for many years to come, even with the spectacular progress in computer technology).
For this reason, the contemporary research in traditional ab initio methods is
concerned mainly with better approximations to a Full CI method, or infinite order
perturbational expansions, which would give good quality reliable results
with reasonable computational effort. Still, the most promising Coupled Cluster (CC)
and Complete Active Space SCF (CASSCF) calculations scale more than 5th power
in molecular size, and are impractical for molecules containing tens of atoms.
X molecules,
the calculations involved in obtaining accurate results are frequently impractical.
We know how to compute it, but we do not have computational power to do it (probably
for many years to come, even with the spectacular progress in computer technology).
For this reason, the contemporary research in traditional ab initio methods is
concerned mainly with better approximations to a Full CI method, or infinite order
perturbational expansions, which would give good quality reliable results
with reasonable computational effort. Still, the most promising Coupled Cluster (CC)
and Complete Active Space SCF (CASSCF) calculations scale more than 5th power
in molecular size, and are impractical for molecules containing tens of atoms.
Density Functional Theory does not provide a prescription how to calculate
truth. It only provides the existence proof for possibility of obtaining
accurate results, but no prescription for systematic improvement. The DFT
is accurate, if we knew how to derive necessary relations between density
and energy. Unfortunately,
energy functionals relating electronic density to energy are unknown,
and there is no general way to improve them beside trying new ones and
judging their quality by the results. As Prof. Perdew once said:
"We are like a blind person who wants to find out how the elephant looks like
by touching its legs" (sorry, the quatation is from memory, and may not
be accurate). But the Density Functional Theory provides a hope for a method
which scales with the size as ![]() in the worst case, and possibly linearly for
larger molecules (Zhou, 1995; Yang, 1991). So it is used in the
spirit of late prof. Slater saying (about his X
in the worst case, and possibly linearly for
larger molecules (Zhou, 1995; Yang, 1991). So it is used in the
spirit of late prof. Slater saying (about his X![]() method): Do you want to
calculate it, or do you want it to be accurate? The DFT results are in many
cases surprisingly good if one takes into account the crude approximations
on which some of them are based. For example, Local Spin Density calculations
yield results on many molecular properties which are of quality comparable
with higher order ab initio correlated methods, yet, the LSD assumes that
electrons in molecules behave like electrons in an uniform electron gas.
Moreover, DFT methods frequently provide reliable answer for cases which are
especially difficult to address with conventional ab initio methods,
like, e.g., transition metals. On the other hand, they frequently fail miserably,
e.g., in charge-transfer complexes.
method): Do you want to
calculate it, or do you want it to be accurate? The DFT results are in many
cases surprisingly good if one takes into account the crude approximations
on which some of them are based. For example, Local Spin Density calculations
yield results on many molecular properties which are of quality comparable
with higher order ab initio correlated methods, yet, the LSD assumes that
electrons in molecules behave like electrons in an uniform electron gas.
Moreover, DFT methods frequently provide reliable answer for cases which are
especially difficult to address with conventional ab initio methods,
like, e.g., transition metals. On the other hand, they frequently fail miserably,
e.g., in charge-transfer complexes.
The fact that more, and more ab initio packages provide options for DFT calculations, is a sign of changing times, and the indication that even the most vigorous opponents of this method see that it is just another way of doing things. On the other hand, DFT is in principle only applicable to the ground state, and there is little hope that it will be extended in a practical way to excited states in a straightforward manner any time soon.
Wave Functions
Since inception of quantum mechanics by Heisenberg, Born, and Jordan
in 1925, and Schrödinger in 1926, there were basically two competing
approaches to find the energy of a system of electrons. One was rooted in
statisticial mechanics and the fundamental variable was the total electron
density ![]() , i.e., the number of electrons per unit volume
at a given point in space (e.g., in cartesian coordinates:
, i.e., the number of electrons per unit volume
at a given point in space (e.g., in cartesian coordinates:
![]() ). In this approach,
electrons were treated as particles forming a special gas, called electron gas.
The special case, the uniform electron gas, corresponds to
the
). In this approach,
electrons were treated as particles forming a special gas, called electron gas.
The special case, the uniform electron gas, corresponds to
the
![]() .
.
Another approach was to derive the many particle
wave function
![]() (where the
(where the ![]() denotes the coordinates of the 1st electron,
denotes the coordinates of the 1st electron,
![]() the 2nd electron, and so on, and
the 2nd electron, and so on, and ![]() is time) and solve the
stationary (time-independent) Schrödinger equation for the system:
is time) and solve the
stationary (time-independent) Schrödinger equation for the system:
| (2) |
Once the function
![]() (or its approximation, i.e., in case
when the Schrödinger equation is
solved only approximately) is known,
the corresponding energy of the system can be calculated
as an expectation value of the hamiltonian
(or its approximation, i.e., in case
when the Schrödinger equation is
solved only approximately) is known,
the corresponding energy of the system can be calculated
as an expectation value of the hamiltonian ![]() , as:
, as:
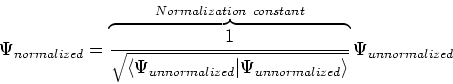
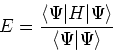 |
(4) |
| (5) |
| (6) |
| (7) |
| (8) |
 |
(9) |
| (10) |
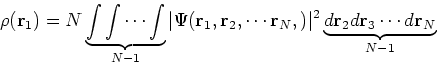 |
(11) |
FUNCTION
Functions is a prescription which maps one or more
numbers to another number.
For example: take a number and multiply it by itself:
![]() , or
take two numbers and add them together:
, or
take two numbers and add them together:
![]() .
Sometimes function does not have a value for some numbers, and only
certain numbers can be used as an argument for a function. E.g., square root
is only defined for nonnegative numbers (if you want to have a real number as
a result).
OPERATOR
.
Sometimes function does not have a value for some numbers, and only
certain numbers can be used as an argument for a function. E.g., square root
is only defined for nonnegative numbers (if you want to have a real number as
a result).
OPERATOR
Operator (usually written with a hat, e.g., ![]() or in
calligraphic style
or in
calligraphic style ![]() ) is
a prescription which maps one function to another function.
For example: take a function and square its value:
) is
a prescription which maps one function to another function.
For example: take a function and square its value:
![]() , e.g.,
, e.g.,
![]() . Or
calculate second derivative of a function versus
. Or
calculate second derivative of a function versus
![]() :
:
![]() , and
, and
![]() .
Nabla is the popular differenial operator in 3 dimensions. In cartesians it is:
.
Nabla is the popular differenial operator in 3 dimensions. In cartesians it is:
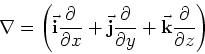
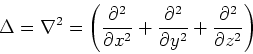
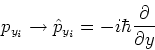
The Schrödinger equation (1) is an example of an eigenproblem, i.e., the
equation in which an operator acts on a function and as a result it returns
the same function multiplied by a constant. For some operators, there are no
nontrivial solutions (trivial means:
![]() ),
but for operators which correspond to some physical
quantity of some physical system, these equations
have solutions in principle, i.e., one can find a set of
functions, and corresponding constants, which satisfy them.
While these eigenproblems
have solutions in principle, these equations may not be
easy to solve. They frequently have the form of partial second order
differential equations, or integro-differential equations, and they
may not be analytically solved in general, some special cases
may have an analytic solution (e.g., one particle, or two particles
which interact in a special way).
),
but for operators which correspond to some physical
quantity of some physical system, these equations
have solutions in principle, i.e., one can find a set of
functions, and corresponding constants, which satisfy them.
While these eigenproblems
have solutions in principle, these equations may not be
easy to solve. They frequently have the form of partial second order
differential equations, or integro-differential equations, and they
may not be analytically solved in general, some special cases
may have an analytic solution (e.g., one particle, or two particles
which interact in a special way).
FUNCTIONAL
Functional takes a function and provides a number. It is usually
written with the function in square brackets as ![]() . For example:
take a function and integrate it from
. For example:
take a function and integrate it from ![]() to
to ![]() :
:
![$F[f] = \int\limits_{-\infty}^{+\infty} f(x) dx$](img72.gif) . Note that the formula for
the expectation value (3) is the total energy functional
. Note that the formula for
the expectation value (3) is the total energy functional
![]() , since it takes some function
, since it takes some function ![]() and returns the value of
energy for this
and returns the value of
energy for this ![]() .
.
Functionals can also have derivatives, which behave similarly to
traditional derivatives for functions. The differential of the functional is
defined as:
![\begin{displaymath}
\delta F[f] = F[f + \delta f] - F[f] = \int \frac{\delta F}{\delta f(x)}
\delta f(x) dx
\end{displaymath}](img74.gif) |
(13) |
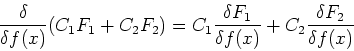 |
(14) |
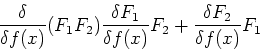 |
(15) |
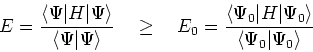 |
(16) |
As said before, we can rarely solve the Schrödinger equation exactly. And we have to use approximations.
The first successful attempt to derive approximate wave functions
for atoms was devised by Hartree (1928).
In this approach the many-electron wave function ![]() is approximated
by the product of one-electron functions
is approximated
by the product of one-electron functions ![]() for each of the
for each of the ![]() electrons:
electrons:
| (18) |
| (19) |
Since nuclei are much heavier than electrons (a proton is
1836 times heavier than an electron),
they have much larger intertia than electrons, and we can consider that electrons will adjust
their positions instantly whenever nuclei move. Prof. Rod Bartlett once used an analogy of
flies around the wedding cakes - you move the cakes, and the flies move with them.
Without much error, we can therefore separate the movement of electrons and nuclei, and
assume that the movement of electrons depends on positions of nuclei in a parametric way.
This is the contents of the Born-Oppenheimer (1927) approximation. It allows us to use the electronic
hamiltonian ![]() to study electrons, and add the components of energy coming from nuclei
(i.e., their kinetic energy,
to study electrons, and add the components of energy coming from nuclei
(i.e., their kinetic energy, ![]() , and internuclear repulsion energy,
, and internuclear repulsion energy, ![]() ) at the
end of calculations.
) at the
end of calculations.
The form of these operators will be given below. Atomic units are used here,
i.e., mass of electron ![]() ;
;
![]() ; length is expressed in bohrs (1 bohr = 0.529177249Å; it is the
radius of the first Bohr orbit in hydrogen atom);
energy is in hartrees (1 hartree is 2 rydbergs, and the ground state energy
of hydrogen atom is
; length is expressed in bohrs (1 bohr = 0.529177249Å; it is the
radius of the first Bohr orbit in hydrogen atom);
energy is in hartrees (1 hartree is 2 rydbergs, and the ground state energy
of hydrogen atom is ![]() rydberg, or 0.5 hartree, 1 hartree = 627.51 kcal/mol = 2625.5 kJ/mol);
1 unit of charge is a charge of a proton, the gaussian electrostatic system is used (the unit
of permittivity is
rydberg, or 0.5 hartree, 1 hartree = 627.51 kcal/mol = 2625.5 kJ/mol);
1 unit of charge is a charge of a proton, the gaussian electrostatic system is used (the unit
of permittivity is
![]() and it does not appear in the Coulomb law for vacuum).
and it does not appear in the Coulomb law for vacuum).
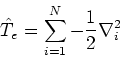 |
(21) |
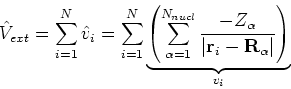 |
(22) |
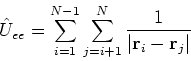 |
(23) |
We can rewrite again the electronic hamiltionian:
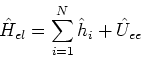 |
(24) |
| (26) |
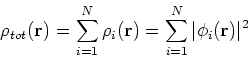 |
(27) |
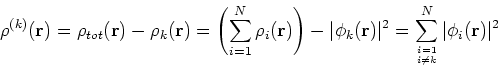 |
(28) |
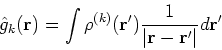 |
(29) |
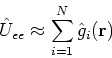 |
(30) |
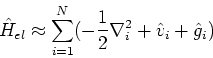 |
(31) |
| (32) |
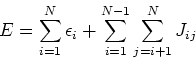 |
(33) |
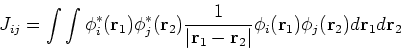 |
(35) |
The Hartree approximation works well for atoms. The one-electron functions are
quite good, and allow us to produce an approximate many-electron function for the whole atom.
But the form of the function adopted by Hartree was basically wrong. It was known
at that time that interchanging the electron labels in the wave functions for the system
of electrons has to change its sign. This is not something which can be derived. It is simply
a law of nature. And it is important, since without this property, the function will not
correctly describe the system. Electrons (fermions) have this propery, and we
should use it. In the system
of fermions, no two particles can be described by the same one particle function.
So, few years later, Fock (1930), and independently Slater(1930) proposed a fix to the
Hartree method. They also used one-electron functions, but the total wave function for the
system was not a simple product of orbitals, but an antisymmetrized sum of all the products
which can be obtained by interchanging electron labels. It is conveniently represented as
a determinant (called Slater determinant):
Let us examine some properties of this function taking a 2-electron case:
![\begin{displaymath}
\Psi({\bf r}_1, {\bf r}_2) = \frac{1}{\sqrt{2}}\left\vert \b...
...\phi_2({\bf r}_2) - \phi_2({\bf r}_1) \phi_1({\bf r}_2)\right]
\end{displaymath}](img148.gif) |
(37) |
If we change electron labels
![]() and
and
![]() we get
we get
![\begin{displaymath}
\Psi({\bf r}_2, {\bf r}_1) = \frac{1}{\sqrt{2}}\left\vert \b...
...\phi_2({\bf r}_1) - \phi_2({\bf r}_2) \phi_1({\bf r}_1)\right]
\end{displaymath}](img151.gif) |
(38) |
![\begin{displaymath}
\Psi({\bf r}_2, {\bf r}_1) = \frac{1}{\sqrt{2}}\left\vert \b...
...f r}_1) - \phi({\bf r}_2) \phi({\bf r}_1)\right]
\equiv {\bf0}
\end{displaymath}](img154.gif) |
(39) |
However, it complicates equations compared to Hartree method and introduces a new term, electron exchange. The method of finding the best single determinant wave function for the system is called Hartree-Fock method.
The expectation value of total energy for the single determinant wave function ![]() is given by:
is given by:
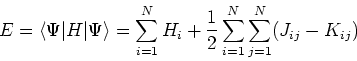 |
(40) |
| (41) |
There are essentially two approaches to HF method: purely numerical,
and the one in which orbitals ![]() are represented as combinations
of basis functions. The numerical HF was used at some
point to derive orbitals for many-electron atoms. There are also some
numerical HF programs for molecules. However, these methods are
quite intensive. It is much more popular to represents orbitals
are represented as combinations
of basis functions. The numerical HF was used at some
point to derive orbitals for many-electron atoms. There are also some
numerical HF programs for molecules. However, these methods are
quite intensive. It is much more popular to represents orbitals ![]() as a linear expansion into a set of some basis functions
as a linear expansion into a set of some basis functions ![]() :
:
The derivation of specific equations for Hartree-Fock
method will not be given
here, but can be found in many books, (see, e.g.:
McWeeny & Sutcliffe, 1969; Parr, 1963; Pilar, 1968;
Slater, 1968; Szabo & Ostlung, 1989) and landmark papers
of Roothaan (1951, 1960).
In the SFC LCAO MO (Self Consistent Field - Linear
Combination of Atomic Orbitals, Molecular Orbitals) method,
we are looking for the coefficients ![]() which minimize the energy.
which minimize the energy.
They are derived using varational principle, i.e.
the goal here is to find a single determinant function ![]() as represented by
equation (36) which corresponds to the lowest possible value of energy.
It is done by the variational calculus. The condition of minimum of energy is:
as represented by
equation (36) which corresponds to the lowest possible value of energy.
It is done by the variational calculus. The condition of minimum of energy is:
| (44) |
| (45) |
The problem of minimization of a function with some subsidiary conditions
is done efficiently with the Lagrange's method of undetermined multipliers.
In this method, the constraints are converted into expressions whose
value is zero when the constraint is met. Such expressions are then
multiplied by an undetermined constant and added (or subtracted, according to
taste) to the minimized function. The resulting sum is then minimized:
| (46) |
As a result, one gets Hartree-Fock eigenequations.
where the Fock operator is defined as:
In these equations, the fact that operators act on certain coordinates is stressed by writing explicitly
their dependence on coordinates of electron 1 (it could be any electron number, since it does not matter
how we number them). The
![]() was defined in equation (25). The Coulomb
operator,
was defined in equation (25). The Coulomb
operator,
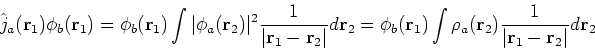 |
(49) |
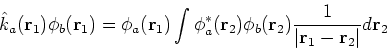 |
(50) |
If we now introduce basis, i.e., replace ![]() 's with their expansions into basis functions
according to equation (43), the eigenproblems become a set of algebraic equations.
Substituting equation (43) into (47) one gets:
's with their expansions into basis functions
according to equation (43), the eigenproblems become a set of algebraic equations.
Substituting equation (43) into (47) one gets:
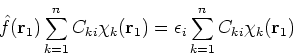 |
(51) |
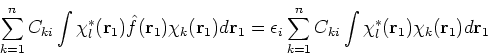 |
(52) |
| (53) |
| (54) |
The problem now is to find such marix ![]() which diagonalizes
which diagonalizes ![]() , which is a standard
procedure in linear algebra. First step is to find a matrix
, which is a standard
procedure in linear algebra. First step is to find a matrix ![]() which diagonalizes overlap integrals
which diagonalizes overlap integrals ![]() , which is a step ortogonalizing the basis set
(i.e., the basis set is converted to linear combinations of original basis functions, which
are mutually orthogonal).
, which is a step ortogonalizing the basis set
(i.e., the basis set is converted to linear combinations of original basis functions, which
are mutually orthogonal).
| (56) |
While they seem simple, the problem is that elements of ![]() depend on orbitals
depend on orbitals ![]() , i.e.,
one can solve them only through iterative process as in Hartree's method. Moreover, due to Coulomb
and exchange operators, the Fock matrix elements involve a massive number of two-electron integrals of a type:
, i.e.,
one can solve them only through iterative process as in Hartree's method. Moreover, due to Coulomb
and exchange operators, the Fock matrix elements involve a massive number of two-electron integrals of a type:
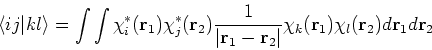 |
(58) |
To escape these difficulties and complexities, people tried for many years to
decribe electron systems using density, rather than wave function.
In the HF approach the two-electron integrals
![]() dominate the computational effort.
Moreover, the HF is an approximation, as it does not account for dynamic correlation
due to the rigid form of single determinant wave function. To solve the HF equations,
the assumption has to be made that electrons interact with the averaged potential coming
from other electrons, while in fact, the interactions between electrons are pairwise.
In reality, electrons correlate their movements trying to avoid each other, so there is least
amount of electrostatic repulsion.
To account for dynamic correlation, one has to go to correlated methods, which use multideterminant
wave functions, and these scale as fifth, or even greater powers with the size of a system.
While HF method is quite successful for geometries, it fails miserably to
describe bond breaking or forming (for review of correlated ab initio methods see: Bartlett & Stanton, 1994).
Density Functional Theory - early developments
dominate the computational effort.
Moreover, the HF is an approximation, as it does not account for dynamic correlation
due to the rigid form of single determinant wave function. To solve the HF equations,
the assumption has to be made that electrons interact with the averaged potential coming
from other electrons, while in fact, the interactions between electrons are pairwise.
In reality, electrons correlate their movements trying to avoid each other, so there is least
amount of electrostatic repulsion.
To account for dynamic correlation, one has to go to correlated methods, which use multideterminant
wave functions, and these scale as fifth, or even greater powers with the size of a system.
While HF method is quite successful for geometries, it fails miserably to
describe bond breaking or forming (for review of correlated ab initio methods see: Bartlett & Stanton, 1994).
Density Functional Theory - early developments
For many years, the use of electron density as a fundamental
description of the system was based on intuition rather than
hard proof that this can be done. Electron density is more
attractive (depends only on ![]() , and eventually, there may be
two densities for spin polarized systems, one for spin up electrons
, and eventually, there may be
two densities for spin polarized systems, one for spin up electrons
![]() and one for spin down electrons
and one for spin down electrons
![]() ,
as opposed to many particle
wave function which depends on all coordinates of all particles, i.e.,
for N electrons, it depends on
,
as opposed to many particle
wave function which depends on all coordinates of all particles, i.e.,
for N electrons, it depends on ![]() variables (or
variables (or ![]() if you count in spin).
The fact that the ground state properties are functionals of the
electron density
if you count in spin).
The fact that the ground state properties are functionals of the
electron density ![]() was proved by Hohenberg and Kohn (1964)
and it provides the basic framework for modern Density Functional
methods.
was proved by Hohenberg and Kohn (1964)
and it provides the basic framework for modern Density Functional
methods.
More specifically, according to the theorem proved by them, the total ground state energy of an electron system can be written as a functional of the electronic density, and this energy is at minimum if the density is an exact density for the groud state. The theorem of HK is an existence proof of such a functional, but there is no prescription how to construct it. If we knew the form of this functional accurately, and if it was not complicated, quantum chemistry would be a done deal. Unfortunately we do not know the exact form of energy functional. It is necessary to use approximations regarding parts of the functional dealing with kinetic energy and exchange and correlation energies of the system of electrons.
The simplest approximation is the local density approximation (LDA) which leads to a Thomas-Fermi (Fermi, 1928; Thomas, 1927) term for kinetic energy (for review see, e.g., Jones & Gunnarsson, 1989; Slater, 1968; March, 1957) and the Dirac (1930)1 term for the exchange energy. The corresponding functional is called Thomas-Fermi-Dirac energy. As you see, these developments are not recent and were parallel to the work done in the wave function approaches. These functionals can be further improved but the results are not that encouraging for molecular systems. But, on the other hand, the Thomas-Fermi-Dirac+improvments method is a true density functional method, since all components of energy are expressed via density alone, without using many particle wave functions. However, for the time being, it seems that there is no way to avoid wave functions in molecular calculations and for accurate calculations they have to be used as a mapping step between the energy and density. For example, the Thomas-Fermii theory does not predict chemical bonds. While "pure" density functional theories are very usefull in studying solid phase (e.g., conductivity), they fail to provide meaningful results for molecular systems.
The predecessor of the modern chemical approaches to the DFT was undoubtely the
Slater's X![]() method formulated in 1951 (Slater, 1951 & 1974, for review see:
Johnson 1973 & 1975). It was
developed as an approximate solution to the HF equations.
In this method, the HF exchange was approximated by:
method formulated in 1951 (Slater, 1951 & 1974, for review see:
Johnson 1973 & 1975). It was
developed as an approximate solution to the HF equations.
In this method, the HF exchange was approximated by:
How these theorems were derived? By quite an original logic. Within a Born-Oppenheimer
approximation, the ground state of the system of electrons is a result of positions of
nuclei. This is obvious, if we look at the ![]() hamiltonian in equation (20).
In this hamiltonian, the kinetic energy of electrons (
hamiltonian in equation (20).
In this hamiltonian, the kinetic energy of electrons (![]() ) and the
electron-electron interaction (
) and the
electron-electron interaction (![]() ) ``adjust'' themselves to the external
(i.e., coming from nuclei) potential
) ``adjust'' themselves to the external
(i.e., coming from nuclei) potential ![]() . Once the
. Once the ![]() is in place,
everything else is, including electron density, which simply adjusts itself to give the
lowest possible total energy of the system. The external potential
is in place,
everything else is, including electron density, which simply adjusts itself to give the
lowest possible total energy of the system. The external potential ![]() is the
only variable term in this equation, and everything else depends indirectly on it.
is the
only variable term in this equation, and everything else depends indirectly on it.
Hohenberg and Kohn posed a more interesting question, which is quite opposite to
the traditional logic: Is ![]() uniquely determined
from the knowledge of electron density
uniquely determined
from the knowledge of electron density ![]() ? Can we find out (in principle,
though it need not be easy) where and what the nuclei are, if we know the density
? Can we find out (in principle,
though it need not be easy) where and what the nuclei are, if we know the density ![]() in the ground state? Is there a precise mapping from
in the ground state? Is there a precise mapping from ![]() to
to ![]() ? The answer to
this question is: Yes!. Actually the mapping is accurate within a constant, which would not
change anything, since Schrödinger equations with
? The answer to
this question is: Yes!. Actually the mapping is accurate within a constant, which would not
change anything, since Schrödinger equations with ![]() and
and
![]() yields
exactly the same eigenfunctions, i.e., states (it is easy to prove based on the linear property
of the hamiltonian), and the energies will be simply elevated by the value of this
yields
exactly the same eigenfunctions, i.e., states (it is easy to prove based on the linear property
of the hamiltonian), and the energies will be simply elevated by the value of this ![]() . Note that
all energies are known only within some constant, which establishes the frame of reference (e.g.,
we do not include electron-Mars gravitational attraction within most calculations).
. Note that
all energies are known only within some constant, which establishes the frame of reference (e.g.,
we do not include electron-Mars gravitational attraction within most calculations).
Why was this question so important? Because, if this is true, the knowledge
of density provides total information about the system, and formally if we know the density,
we know everything there is to known.
Since ![]() determines number of electrons,
determines number of electrons, ![]() :
:
| (60) |
| (63) |
| (64) |
Additionally, HK grouped together all functionals
which are secondary (i.e., which are responses)
to the ![]() :
:
The second HK theorem provides variational principle in electron density representation ![]() 3.
For a trial density
3.
For a trial density
![]() such that
such that
![]() and for which
and for which
![]() ,
,
| (66) |
As to the necessary conditions for this theorem, there is still some controversy concerning
the, so called, representability of density. The ![]() -representability, i.e., the fact that the trial
-representability, i.e., the fact that the trial ![]() has to
sum up to
has to
sum up to ![]() electrons is easy to achieve by simple rescaling. It is
automatically insured if
electrons is easy to achieve by simple rescaling. It is
automatically insured if ![]() can be mapped to some wave function
(for further discussion see: Parr & Yang, 1989; Gilbert, 1975; Lieb, 1982;
and Harriman, 1980).
Assuring that the trial density has also
can be mapped to some wave function
(for further discussion see: Parr & Yang, 1989; Gilbert, 1975; Lieb, 1982;
and Harriman, 1980).
Assuring that the trial density has also ![]() -representability
(usually denoted in the literature as
-representability
(usually denoted in the literature as ![]() -representability) is not that easy. Levy (1982) and Lieb (1983)
have shown, that there are some ``reasonable'' trial densities, which are not the ground state densities for any
possible
-representability) is not that easy. Levy (1982) and Lieb (1983)
have shown, that there are some ``reasonable'' trial densities, which are not the ground state densities for any
possible ![]() potential, i.e., they do not map to any external potential. Such densities do not correspond therefore
to any ground state, and their optimization will not lead to a ground state.
Moreover, during energy minimization, we may take a wrong turn, and get
stuck into some non
potential, i.e., they do not map to any external potential. Such densities do not correspond therefore
to any ground state, and their optimization will not lead to a ground state.
Moreover, during energy minimization, we may take a wrong turn, and get
stuck into some non ![]() -representable density and never be able to converged
to a physically relevant ground state density.
For an interesting discussion, see
Hohenberg et al. (1990). Assuming that we restrict ourselved only to
trial densities which are both
-representable density and never be able to converged
to a physically relevant ground state density.
For an interesting discussion, see
Hohenberg et al. (1990). Assuming that we restrict ourselved only to
trial densities which are both ![]() and
and ![]() representable, the variational principle for density is easly proven, since
each trial density
representable, the variational principle for density is easly proven, since
each trial density ![]() defines a hamiltonian
defines a hamiltonian
![]() . From the hamiltonian
we can derive the corresponding wave function
. From the hamiltonian
we can derive the corresponding wave function ![]() for the ground state represented by this hamiltonian. And
according to the traditional variational principle, this wave function
for the ground state represented by this hamiltonian. And
according to the traditional variational principle, this wave function ![]() will not be a ground state
for the hamiltonian of the real system
will not be a ground state
for the hamiltonian of the real system ![]() :
:
| (67) |
The condition of minimum for the energy functional:
![]() needs to be constrained
by the
needs to be constrained
by the ![]() -representability of density which is optimized4.
The Lagrange's method of undetermined multipliers is a very
convenient approach for the constrained minimization
problems. In this method
we represent constraints in such a way that their value is
exactly zero when they are satisfied. In our case, the
-representability of density which is optimized4.
The Lagrange's method of undetermined multipliers is a very
convenient approach for the constrained minimization
problems. In this method
we represent constraints in such a way that their value is
exactly zero when they are satisfied. In our case, the
![]() representability constraint can be represented as:
representability constraint can be represented as:
| (68) |
| (69) |
| (70) |
| (71) |
![\begin{displaymath}
F[f + \delta f] - F[f] = \delta F= \int \frac{\delta F}{\delta f(x)}\delta f(x) dx
\end{displaymath}](img242.gif) |
(72) |
![\begin{displaymath}
\int \frac{\delta E[\rho({\bf r})]}{\delta \rho({\bf r})}\de...
...f r}) d {\bf r}
- \mu \int \delta \rho({\bf r}) d {\bf r} = 0
\end{displaymath}](img243.gif) |
(73) |
![\begin{displaymath}
\int \left\{ \frac{\delta E[\rho({\bf r})]}{\delta \rho({\bf r})}
- \mu \right\} \delta \rho({\bf r}) d {\bf r} = 0
\end{displaymath}](img244.gif) |
(74) |
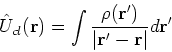 |
(77) |
![\begin{displaymath}
E_{cl}[\rho] = \int \int \frac{\rho({\bf r}^\prime) \rho({\b...
...}{\vert{\bf r}^\prime - {\bf r}\vert}d {\bf r} d{\bf r}^\prime
\end{displaymath}](img249.gif) |
(78) |
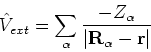 |
(79) |
![\begin{displaymath}
\mu = \frac{\delta E[\rho({\bf r})]}{\delta \rho({\bf r})} =...
...}) + \frac{\delta E_{xc}[\rho({\bf r})]}{\delta \rho({\bf r})}
\end{displaymath}](img254.gif) |
(80) |
| (82) |
![\begin{displaymath}
\hat{V}_{xc}({\bf r}) = \frac{\delta E_{xc}[\rho({\bf r})]}{\delta \rho({\bf r})}
\end{displaymath}](img257.gif) |
(83) |
| (84) |
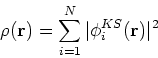 |
(85) |
![\begin{displaymath}
T_0[\rho] = \frac{1}{2} \sum_{i=1}^N \left<\phi_i^{KS}\vert\nabla_i^2\vert\phi_i^{KS}\right>
\end{displaymath}](img263.gif) |
(86) |
| (87) |
![\begin{displaymath}
E[\rho] = \sum_{i=1}^{N}\epsilon_i - \frac{1}{2} \int \int \...
...nt \hat{V}_{xc}({\bf r})\rho({\bf r}) d {\bf r} + E_{xc}[\rho]
\end{displaymath}](img265.gif) |
(88) |
| (89) |
First implementations of the Kohn-Sham method were using the local approximations to the exchange correlation energy.
The appropriate functionals were taken from data on homogenous electron gas.
There were two variants of the method: spin unpolarized (LDF/LDA - Local Density Functional/Approximation)
and spin polarized (LSD - Local Spin Density) where arguments require both ![]() and
and ![]() electron
densities, rather than
a total density.
electron
densities, rather than
a total density.
For historical reasons, the exchange correlation energy was partitioned into 2 parts:
| (90) |
| (91) |
Early attempts to improve functionals by GEA (Gradient Expansion Approximation), in which ![]() was
expanded in Taylor series versus
was
expanded in Taylor series versus ![]() and truncated at a linear term, did not improve results too much (see, e.g.
Langreth & Vosko, 1990). Only GGA (Generalized Gradient Approximation) provided notable improvements by
expanding
and truncated at a linear term, did not improve results too much (see, e.g.
Langreth & Vosko, 1990). Only GGA (Generalized Gradient Approximation) provided notable improvements by
expanding ![]() . The expansion here is not a simple Taylor expansion, but tries
to find the right asymptotic behaviour and right scaling for the
usually nonlinear expansion. These enhanced functionals are frequently called nonlocal or gradient corrections,
since they depend not only upon the density, but also the magnitude of the gradient (i.e., first derivative)
of density at a given point.
While the term corrections has a historical meaning, where the corrections were added on the top of local
density functionals, it is probably no longer correct, since modern nonlocal functionals are quite complicated
functions in which the value of density and its gradient are integral parts of the formula. There are many different
corrections, and it will take some time when their respective merit, accuracy, and best domains of applications will be
established. For review of the existing functionals see e.g.: Clementi (1994), Johnson et al (1993),
Seminario & Politzer (1995), Ziegler (1991). The most widely used local potentials are: Slater for exchange (Slater, 1974),
and VWN for correlation (Vosko et al, 1980).
Probably the most frequently in use today are:
. The expansion here is not a simple Taylor expansion, but tries
to find the right asymptotic behaviour and right scaling for the
usually nonlinear expansion. These enhanced functionals are frequently called nonlocal or gradient corrections,
since they depend not only upon the density, but also the magnitude of the gradient (i.e., first derivative)
of density at a given point.
While the term corrections has a historical meaning, where the corrections were added on the top of local
density functionals, it is probably no longer correct, since modern nonlocal functionals are quite complicated
functions in which the value of density and its gradient are integral parts of the formula. There are many different
corrections, and it will take some time when their respective merit, accuracy, and best domains of applications will be
established. For review of the existing functionals see e.g.: Clementi (1994), Johnson et al (1993),
Seminario & Politzer (1995), Ziegler (1991). The most widely used local potentials are: Slater for exchange (Slater, 1974),
and VWN for correlation (Vosko et al, 1980).
Probably the most frequently in use today are:
Beside different exchange and correlation functionals, there is recent interest in the
ACM (Adiabatic Connection Method) (see: Harris, 1984; Becke, 1993) to include at least
partially the exact exchange from Hartree-Fock type calculation, or exchange
Kohn-Sham orbitals. This method looks very promising
but its application is not without cost. In the worst case, ACM calculation of
exact exchange via exchange integrals (see equation 42) scales as ![]() .
.
Most molecular DFT codes use basis functions, with a notable exception of
a totally basis free NUMOL (Becke & Dickinson, 1990) program. Quite impressive
variety of basis sets is used. Since most energy components have to be
calculated via numerical integration, the use of contracted
gaussians as a basis is not that essential. But most program use them,
as it allows a substantial code reuse from the well developed traditional
ab initio techniques. The well known deMon (Salahub et al, 1992), , DGauss
(Andzelm & Wimmer, 1992), and DeFT (a program similar to deMon written aby Alain St-Amant et al.
and available at
ftp://www.ccl.net/pub/chemistry/software/SOURCES/FORTRAN/DeFT)
all use gaussian basis functions. Some traditional ab initio packages also
include options to perform DFT calculations: ACES2 (Bartlett & Stanton, 1994),
Cadpac5 (Cambridge Analytic Derivative Package, e.g., Murray et al, 1993), Gaussian (Gaussian, Inc.),
Spartan (Wavefunction, Inc.), and I hear that many others.
The AMOL, recently renamed to ADF (Amsterdam Density Functional) (Boerrigter et al 1998) uses Slater type basis.
The DVM (Ellis & Painter, 1970) and DMol (Delley, 1995) use numerical basis sets, which are given as spline functions for radial part (the angular part is taken as appropriate spherical harmonics).
There are also programs for extended systems like: Corning (Teter et al, 1989),
Crystal94 (Univ. Turin & SERC Daresbury Lab), and Wien95 (Blaha et al, 1995)
which use LAPW (Linearized Augmented Plane Wave's) as basis functions.
NOTE: This stuff was written in 1996 - there are many new
codes and new versions out there
Typical Program Organization for SCF-KS equations
The single geometry SCF cycle or geometry optimization
involve following steps:
It is quite popular to limit expense of numerical integration during the SCF cycle.
It is frequently done by fitting auxiliary functions to charge density and exchange
correlation potential. This allows for much faster integral evaluation.
These auxiliary fitting functions are usually uncontracted gaussians (though quite different
from the atomic basis sets) for which the integrals required for KS matrix can be
calculated analytically. Different auxilliary sets are used for fitting charge density
and exchange-correlation potential (see e.g., Dunlap & Rösch, 1990). The need for
fitting is recently questioned (see e.g., Johnson, 1995) since it scales
as ![]() even for very large systems, however, it is still very popular in
DFT codes. The fitting procedures are in general non sparse, while for large
molecules many contributions coming from distant portions may be neglected
leading to less steep scaling with molecular size.
even for very large systems, however, it is still very popular in
DFT codes. The fitting procedures are in general non sparse, while for large
molecules many contributions coming from distant portions may be neglected
leading to less steep scaling with molecular size.
Early DFT codes were impaired by the lack of analytical gradients. Currently, expressions for first and second derivatives exist (see e.g.: Dunlap & Andzelm, Komornicki & Fitzgerald, 1993) and are implemented in many programs, thus facilitating geometry optimization and vibrational frequency calculations.
Performance of DFT
This will be a very short list of DFT applications, as there are many excellent reviews on this topic
(see e.g.: Labanowski & Andzelm, 1991; Parr & Yang, 1989; Seminario & Politzer 1995; Ziegler, 1991; Bartolotti and Flurchick, 1996; St-Amant. 1996).
The G1 database of Pople and coworkers is a benchmark for accuracy of the traditional ab initio methods.
The database containes 55 molecules for which experimental values of atomization energies are known within
1 kcal/mol. Curtiss et al (1991) achieved the 1.2 kcal/mol mean absolute error for these 55 atomization
energies using the G2 procedure, which is a quite involved prescription incorporating higher order correlated methods.
Becke (1992) was able to reproduce values in this database with a mean absolute error of 3.7 kcal/mol using his NUMOL
program with
gradient corrected functionals. This result was additionally improved by Becke (1993) to 2.4 kcal/mol when portion of
the electron exchange entering the echange-correlation energy, ![]() was calculated exactly form Kohn-Sham orbitals
While the error in DFT is considered still too big, these results were obtained with a method
which is substantially less computationally demanding than
original correlated ab initio procedures used by Pople and coworkers. Obviusly, the errors refer to absolute
atomization energies, which in general are very difficult to calculate with good occuracy
(for review see, e.g., Hehre et al, 1986). The relative differences are usually reproduced much better
with DFT methods.
was calculated exactly form Kohn-Sham orbitals
While the error in DFT is considered still too big, these results were obtained with a method
which is substantially less computationally demanding than
original correlated ab initio procedures used by Pople and coworkers. Obviusly, the errors refer to absolute
atomization energies, which in general are very difficult to calculate with good occuracy
(for review see, e.g., Hehre et al, 1986). The relative differences are usually reproduced much better
with DFT methods.
Even without gradient corrections DFT results for bond dissociation energies are usually much better then the Hartree-Fock (which routinely underbinds) results, though they have an overbinding tendency. The LDA results are approximately of MP2 quality. The inclusion of gradient corrections to DFT provides bond dissociaction energies which pair in occuracy with MP4 and CC results.
Molecular geometries even with LSD are much better than corresponding HF results and are of the MP2 quality. However, LSD fails to correctly treat hydrogen bonding. This deficiency is removed when one uses gradient corrected DFT approaches. Quite estonishingly, DFT provides excellent results for molecules which are notoriously difficult for traditional ab initio approaches like FOOF, FON, and metalorganic or inorganic moities. There seems to be a funny regularity: "If something does not work with ab initio, try it with DFT, and vice versa".
Transition states of organic molecules are frequently not reproduced well with "pure" DFT. However, it seems that admixture of exact exchange (see Becke, 1993) via ACM dramatically improves the problem cases. DFT is however a metod of choice for transition states in metalorganic reactions. These systems are notoriously difficult to treat with even high quality ab initio and have problems with convergence.
Vibrational frequencies are well reproduced even by LSD, though gradient corrections improve agreement with experiment even further.
Ionization potentials, electron affinities, and proton affinities are reproduced fairly well within gradient corrected DFT.
Recently, there is much interest in using DFT for high spin species, since preliminary results are very promissing. On the other hand good performance of DFT in this field comes as a surprise, since high multiplets are poorly described by a single determinant wave function. For all the wrong reasons, with KS wave function of broken symmetry, the multiplets splitting are quite well reproduced by DFT.
The scope of applcations for DFT grows rapidly with calculations of new molecular properties being added to actively developed software. Recent extensions include parameters for NMR and ESR spectroscopy, diamagnetic properties, polarizabilities, relativistic calculations, and others.
The DFT methodology is still far from mature. It is encouraging that several groups which were primarily focused on the development of traditional ab initio approaches are now actively working in the DFT area. It will bring new methodological developments, as well as, more carefull assesment of applicability. With all the hopes which DFT brings, one has to keep in mind, that it is primarily a ground state oriented method, and cannot compete with semiempirical and correlated ab initio methods in calculations concerning excited states.
Advantages of DFT in biological chemistry
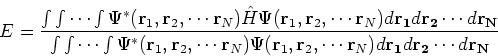
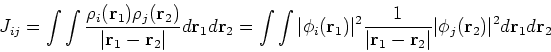
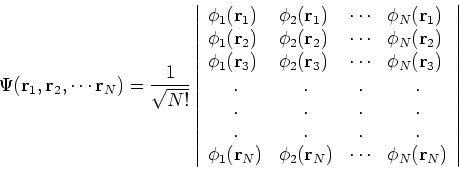
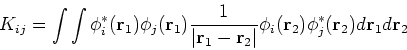
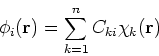
![\begin{displaymath}
\hat{f}({\bf r}_1) = \hat{h}({\bf r}_1) + \sum_{a=1}^N\left[\hat{j}_a({\bf r}_1) - \hat{k}_a({\bf r}_1)\right]
\end{displaymath}](img173.gif)
![\begin{displaymath}
E_{X\alpha}[\rho_\uparrow, \rho_\downarrow]
= -\frac{9}{4}\...
...}{3}({\bf r})
+ \rho_\downarrow^\frac{4}{3}({\bf r})]d {\bf r}
\end{displaymath}](img200.gif)
![\begin{displaymath}
E_0 < \left<\Psi^\prime\vert H\vert\Psi^\prime\right> =
\ove...
...t \rho({\bf r})[\hat{V}_{ext} - \hat{V}_{ext}^\prime]d {\bf r}
\end{displaymath}](img213.gif)
![\begin{displaymath}
E_0^\prime < \left<\Psi\vert H^\prime\vert\Psi\right> = \und...
...t \rho({\bf r})[\hat{V}_{ext} - \hat{V}_{ext}^\prime]d {\bf r}
\end{displaymath}](img215.gif)
![\begin{displaymath}
\mu = \frac{\delta E[\rho({\bf r})]}{\delta \rho({\bf r})}
=...
...r}) + \frac{\delta F_{HK}(\rho({\bf r})}{\delta \rho({\bf r})}
\end{displaymath}](img245.gif)
![\begin{displaymath}
\mu = \frac{\delta E[\rho({\bf r})]}{\delta \rho({\bf r})} =...
...\rho({\bf r})]}{\delta \rho({\bf r})} +
\hat{V}_{eff}({\bf r})
\end{displaymath}](img255.gif)