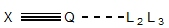|
Introduction
Scaffold hopping is an approach used to discover new chemical classes by
replacing a
portion (the scaffold) of a known compound, while preserving the remaining
chemical groups, under the assumption that they are important for biological
activity. For example, in peptidomimetic efforts, certain peptide sidechains
are the groups that are to be preserved during a search for a new linker (the
scaffold) that preserves the presentation of the sidechain groups
with the receptor. In many cases, the scaffold is a ring system whose
substituents are the groups that must be preserved:

In principle, 2D methods such as substructure or similarity search
can be used, however, scaffold hopping generally is most successful when
attempted with 3D methods.
CAVEAT [Lauri 1994] is one of the early programs developed for
3D scaffold replacement. The CAVEAT methodology is based on specifying two
or more bonds linking the scaffold and R-groups in a 3D bioactive conformer of
a lead compound (the query bonds). These bonds define 3D vectors in
which the origin of the vector is an atom on the existing scaffold and the
terminus of the vector is an atom of the R-group to be preserved. A 3D
database of candidate scaffold conformations is searched for molecules that,
if suitably substituted, would coincide with the substitution bonds of the defined
vectors. The 3D scaffold database contains conformations of molecules as
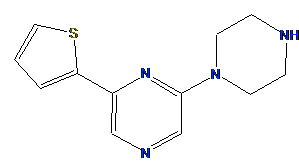
well as annotations that encode the location of potential substituents. A sample linker is shown in Figure 1.
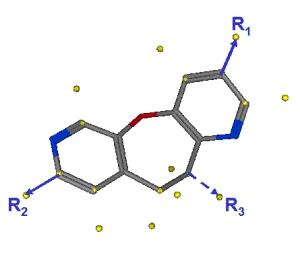
Figure 1.
A 3D linker conformation with potential exit vectors.
By overlaying the potential R-group bonds from the scaffold database molecules
with the defined query bonds, the bond lengths, angles, and orientation of the
original R-groups are preserved. If the bonds overlay sufficiently well, then
a potential new scaffold will have been found. The advantages of the
CAVEAT methodology are that a) the queries are easily specified (select
two scaffold to R-group bonds called the link bonds)
and b) the search can be performed rapidly (overlay vector segments in 3D).
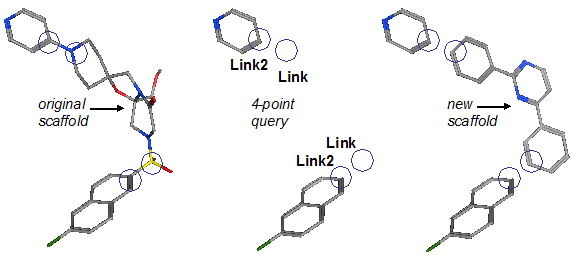
Figure 2. Sample scaffold replacement query. The original ligand has two
R-groups defined. A pair of (Link,Link2)
features are used to preserve each bond. Searching a database of candidate
linkers gives new potential replacement scaffolds.
The original CAVEAT databases consisted of hydrocarbon ring systems or small
alkane substituents of small ring systems, although in principle, any
database can be searched. Vendor catalogs can be preprocessed by removing
“decorations” (e.g., with RECAP [Lewell 1998] rules) and retaining the
unique fragments which are then subjected to conformational search.
Alternatively, structural databases such as the CSD can be similarly processed
to produce 3D fragments more directly.
A more recent method is called Recore [Maass 2007]. Like CAVEAT, Recore
requires the definition of at least two exit vectors, but this approach
allows additional pharmacophore-type features or constraints that a candidate
scaffold must satisfy — for example, the existence of a hydrogen bond
acceptor at a specific location in space. Recore rapidly searches a specially
indexed 3D database although the principles are the same as those of CAVEAT
which
conducts a linear search. The addition of pharmacophore constraints or
filters on the output, however, increases utility of the method.
The combination of CAVEAT methodology with pharmacophore-type methodologies provides a
key advantage over the more structurally sensitive 2D methods.
�Specifically, the important interactions provided by the scaffold can be
preserved while retaining the required geometry of the attached R-groups. The resulting re-scaffolded
lead is then more likely to be a viable new chemical direction for lead
optimization.
In a structure-based context (where the starting bioactive conformation is
often obtained) further constraints can be imposed. The receptor volume
can be used as a shape guide to eliminate candidate scaffolds that would
clash with the receptor. In this article, we present the MOE scaffold
replacement method which combines the CAVEAT methodology with full pharmacophore
discovery capabilities as well as the ability to include structure-based
information, definition of chemistry rules through SMARTS patterns as well
as efficient database searching. We believe that these enhancements are a
significant advancement in 3D scaffold replacement techniques.
The fundamental approach is to combine CAVEAT style functionality with the
MOE pharmacophore tools. Specifically, we will define the substituent
vectors with special pharmacophore annotations called Link annotations.
Link-type annotations denote points of substitution on a candidate scaffold
molecule as well as the locations of potential R-group substituents.
For example, in Figure 1,
the yellow spheres are the Link annotations. Some are placed on
the heavy atoms of the molecule while others are projected away from the molecule.
The heavy atom annotations are the possible substitution points and the
projected annotations are placed at bonded distances and angles consistent
with the hybridization of the heavy atom. The idea is that one Link-type
annotation is placed at the origin of a CAVEAT vector and one at the vector terminus.
During a pharmacophore search, a constraint is imposed that
ensures that both heavy and projected annotations match simultaneously or
not at all. In this way, a pair of Link-type annotations simulates the
CAVEAT “exit vector”.
There are three types of link annotations:
- Link:
annotates a scaffold heavy atom substitution point with at least one (implicit) hydrogen.
- Link2:
annotates projected locations of potential sp2 R-groups.
- Link3:
annotates projected locations of potential sp3 R-groups.
A typical query will consist of (Link,Link2)
or (Link,Link3) pairs located at positions
intended to represent scaffold / R-group bonds. The Link2
or Link3 feature is placed on an existing R-group heavy
atom (~1.5 Å away from the corresponding scaffold atom) and the
Link feature is intended to match a heavy atom of a new
scaffold to be found in a database of scaffold or linker molecules (see
Figure 1). A matching scaffold is then likely to present the existing
R-group in the proper direction.
Link2 query features are used when the R-group atom is sp2.
Link3 query features are used when the R-group atom is
sp3. For example, when attempting to
match a piperazine scaffold with an aromatic R-group, a Link2
query feature should be used so that the piperazine nitrogen conjugated planar
geometry is used (and not the nitrogen tetrahedral geometry).
Scaffold molecules are annotated by an automatic procedure.
Only C, N, O, S, and P atoms with 1, 2, or 3 heavy neighbors and at least one
(possibly implicit) hydrogen are candidates for Link
features; other atoms are not considered as candidates. The following rules
assume that the foregoing conditions are satisfied. Not all these atoms will
be given Link annotations; substituents on freely rotatable
single bonds will be avoided (e.g., sp2-sp3
rotors).
| 
| 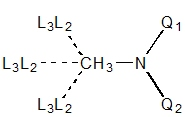
|
| Q = any 1°, Xi = any
| Q = 1° {C,N+}, Y = 2° {O,S,N-}
|
N = 2° or 3°, Q1 = 4-coord, Q2 = any
|
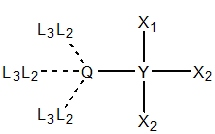
| 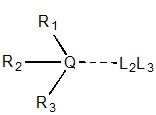
|
| Y ≠ 1°, Q = {OH, SH, NH2, PH2}
|
Q = {C,N,P,S}, Q = 3° sp3,
Ri ≠ H
|
For primary sp3 centers with one substituent
(e.g., 1,1,1-trichloroethane), there are three potential exit vectors
in a tetrahedral geometry. Each point is annotated with Link2&Link3 since
the local geometry is not affected by a substituent's hybridization. A similar
rule is used for tertiary sp3 centers where there is only
one exit vector to preserve the tetrahedral geometry.
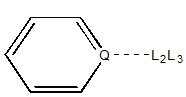
| 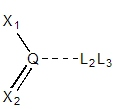
| 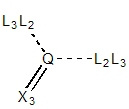
|
| Q = 2° aromatic
| Q = 2° sp2
| Q = 1° sp2
|
sp2 centers have exit vectors to retain the trigonal
planar geometry. For example, substitution on an aromatic nitrogen
(e.g., pyridine) will happen at 120° from the ring atoms
regardless whether the substituent connected atom is sp2
or sp3 hybridized. Other secondary and primary
sp2 centers will be annotated with one and two exit
vector, respectively, retaining the trigonal planar geometry.
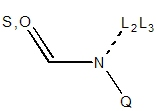
| 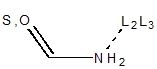
| 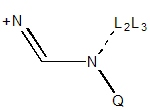
|
| N = 2° amide not in 3,4-ring
| N = 1° amide not in 3,4-ring
| N = 2° in NCN+ not in 3,4-ring
|
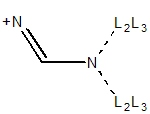
|
| N = 1° in NCN+ not in 3,4-ring
|
Secondary sp3 centers have exit vectors to retain tetrahedral
geometry. However, an extra exit vector is added if a bond is
formed with an sp2 center, resulting in a trigonal geometry.
For primary and secondary amides, the geometry on the nitrogen must be trigonal
planar because of the delocalization. For a primary amide, two exit vectors are
defined, and for a secondary amide one exit vector is defined at 120° to
retain the trigonal planar geometry. Similarly, a primary NCN+ will have two
possible exit vectors while a secondary NCN+ group will have one retaining
the trigonal planar geometry. These rules are independent of the hybridization
of the connected substituent atom. Also, note that cis peptide formation is avoided.
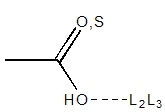
| 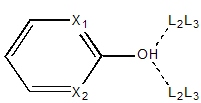
|
| O = 1°
| OH on Caro, Xi = 2°
|
Carboxylic acids have a single exit vector defined at 120° from the C-O-H
center. Any hydroxyl group
substituted on an aromatic ring (e.g., phenol) will have two exit
vectors forming a 120° trigonal planar center ensuring coplanarity with
the ring atoms.
sp centers only have one possible exit vector defined to be colinear
with the triple bond.
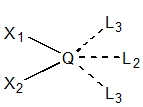
|
|
|
Q = 2° {C,N} sp3, Xi ≠ H
|
|
A secondary amine can be used to show the difference between the geometry of
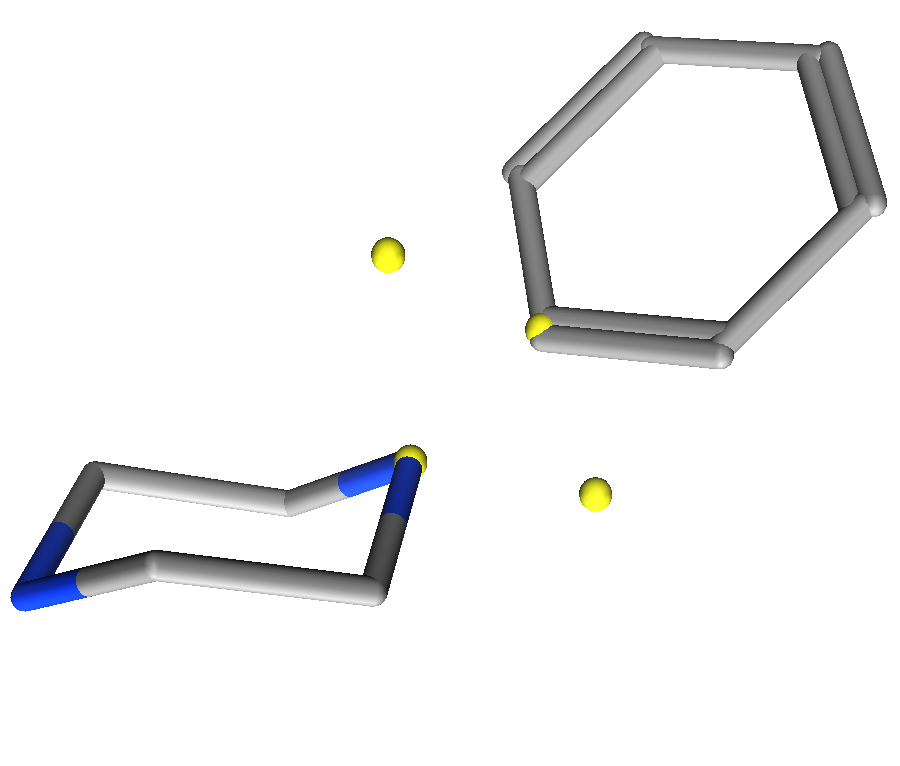
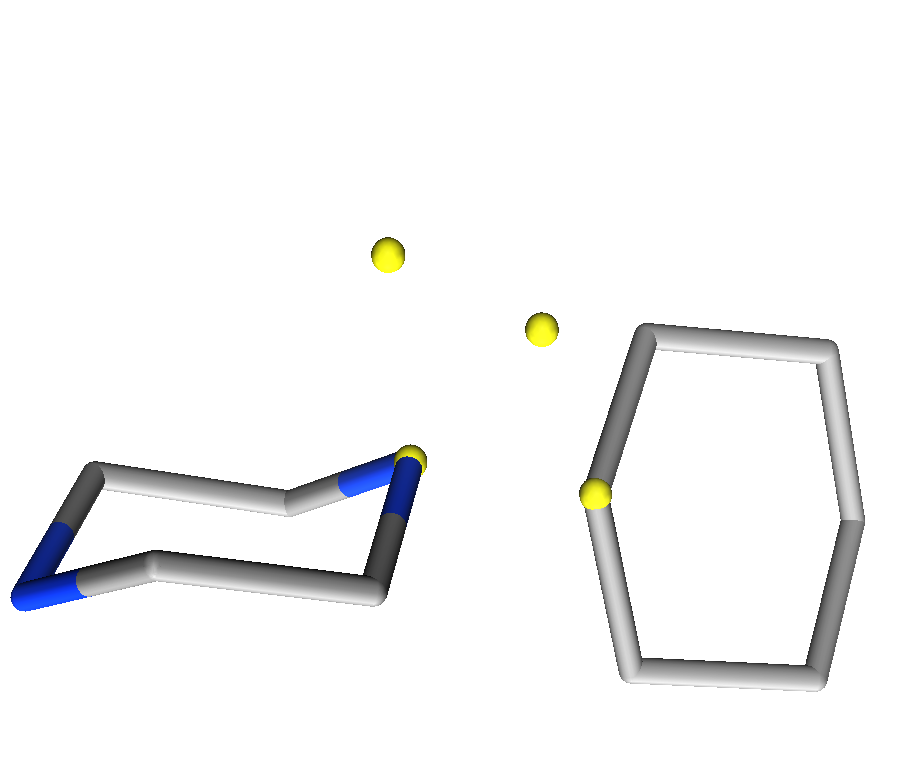
Link2 and Link3. As shown in the figure below,
each nitrogen can adopt either a tetrahedral geometry (Link3)
or extend the π plane of the connected substituent. In the case of
piperazine, an exit vector for substitution of cyclohexane would require a
Link3 feature, while an exit vector for substitution of
benzene would require a Link2 feature. The
Link2 feature would enforce a flat nitrogen extending
the π plane of the benzene ring.
Figure 3: Differentiation between Link2 and Link3 annotations. Left) benzene substitution for Link2 annotations Right) cyclohexane subsitution for Link3 in a tetrahedral geometry.
Note that in most cases, Link2&Link3
annotations are used for the projected annotations. For 2° nitrogen and
carbon atoms, there is a distinction between Link2 and
Link3. Link2 projections are in the
potential π system plane and Link3 projections are in
tetrahedral formation.
The general strategy for scaffold replacement is therefore:
-
Obtain the structure of the active site and a template ligand.
-
Identify
substituent locations from expert knowledge of the system or with
the help active site analysis tools such as ligand interaction
diagrams, electrostatic maps, or contact statistics.
-
Assign special
pharmacophore features to preserve the bonds that link the scaffold
with each substituent.
-
Add excluded volumes to avoid clashes with the receptor and the substituents.
-
Include any additional pharmacophore feature(s) from key interaction(s) present in
the scaffold of the native ligand.
-
Search a database of candidate linkers and select replacement scaffolds.
- Connect the ligand and the R-group(s) to form the new molecule.
The most reliable pharmacophore information can be obtained from high
resolution crystal structures or high-quality docking results. Here,
we will examine Factor Xa in complex with the M55532 ligand (PDB:1IOE).
Factor Xa is a vitamin-K-dependent serine protease that is responsible
for the generation of thrombin from prothrombin in the coagulation cascade.
As a result, Factor Xa is an important target for the development
of anticoagulant drugs [Davie 1991] [Kastenholz 2000].
Before creating special pharmacophore queries for scaffold replacement,
one must first identify the R-groups that are to be preserved.
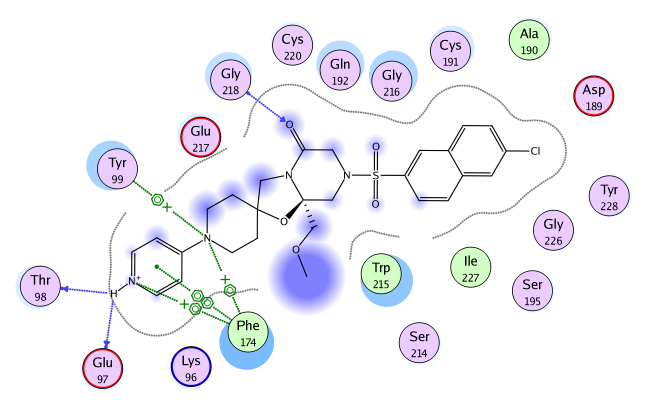
Figure 4.
Ligand Interaction Diagram for Factor Xa (IIOE). The active site
residues are represented as follows: polar residues in pink, hydrophobic
residues in green, acidic residues with a red contour ring, basic
residues with a blue contour ring. Green and blue arrows indicate
hydrogen bonding to sidechain and backbone atoms respectively.
A naphthyl icon represents a π-π stacking interaction, while a benzene with a +
represents a cation-π interaction.
Blue “clouds” on ligand
atoms indicate the solvent exposed surface area of ligand atoms
(darker and larger clouds means more solvent exposure).
Light-blue “halos” around residues indicate the degree of
interaction with ligand atoms (larger, darker halos means more
interaction). The dotted contour reflects steric room for methyl
substitution. The contour line is broken if it is closest to an atom
which is fully exposed.
The ligand interaction diagram [Clark 2007] in Figure 4 shows important interactions
between the M55532 ligand and Factor Xa.
The 4-amino pyridine group (in the hydrophobic D pocket) forms backbone
hydrogen bond interactions with Thr98 and Glu97 as well as cation-π
interactions with Phe174 and Thr99. The 4-amino pyridine group also
forms a π-π stacking interaction with Phe174,
which accommodates hydrophobic and positively charged functional
groups [Stubbs 1995].
At the entrance of the S1 pocket, the Gln192 and Gly216 residues have a
strong hydrophobic interaction with the ligand (as indicated by the large,
dark halo around the residue in Figure 4). A hydrogen bond is also
formed between the lactam carbonyl oxygen of M55532 and
backbone nitrogen of Gly218.
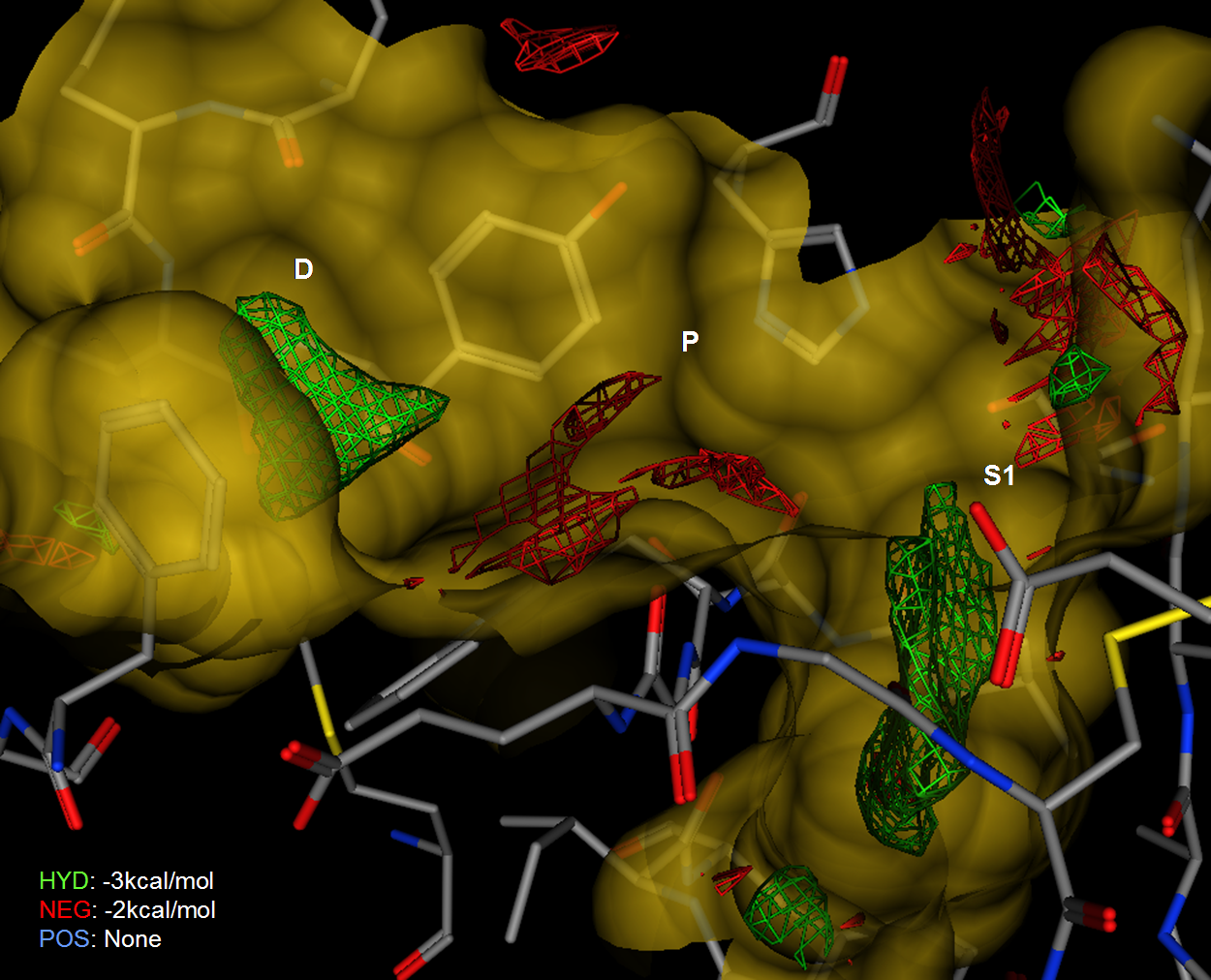
Figure 5.
An Electrostatic Map of Factor Xa calculated from the receptor
structure without the ligand. Negative preferences in red drawn at
~2 kcal/mol and neutral in green plotted at 3kcal/mol.
Figure 5 shows electrostatic isocontours in the active site of
Factor Xa (1IOE) along with a re-entrant surface and pocket labels.
The electrostatic isocontours are drawn by Electrostatic Maps which is an
implementation of a non-linear Poisson-Boltzmann equation solver to the prediction of
electrostatically preferred locations of hydrophobic, negative and positive
regions in a receptor active site.
The Factor Xa structure was prepared for electrostatic analysis by
assigning standard ionization states, adding protons to satisfy valence requirements
and calculating partial charges using the MMFF94 forcefield [Halgren 1996].
In Figure 5, the green contour shows the hydrophobic regions and the red
contour shows the negative regions. The positive regions of the
electrostatic maps was minimal and is not displayed for clarity.
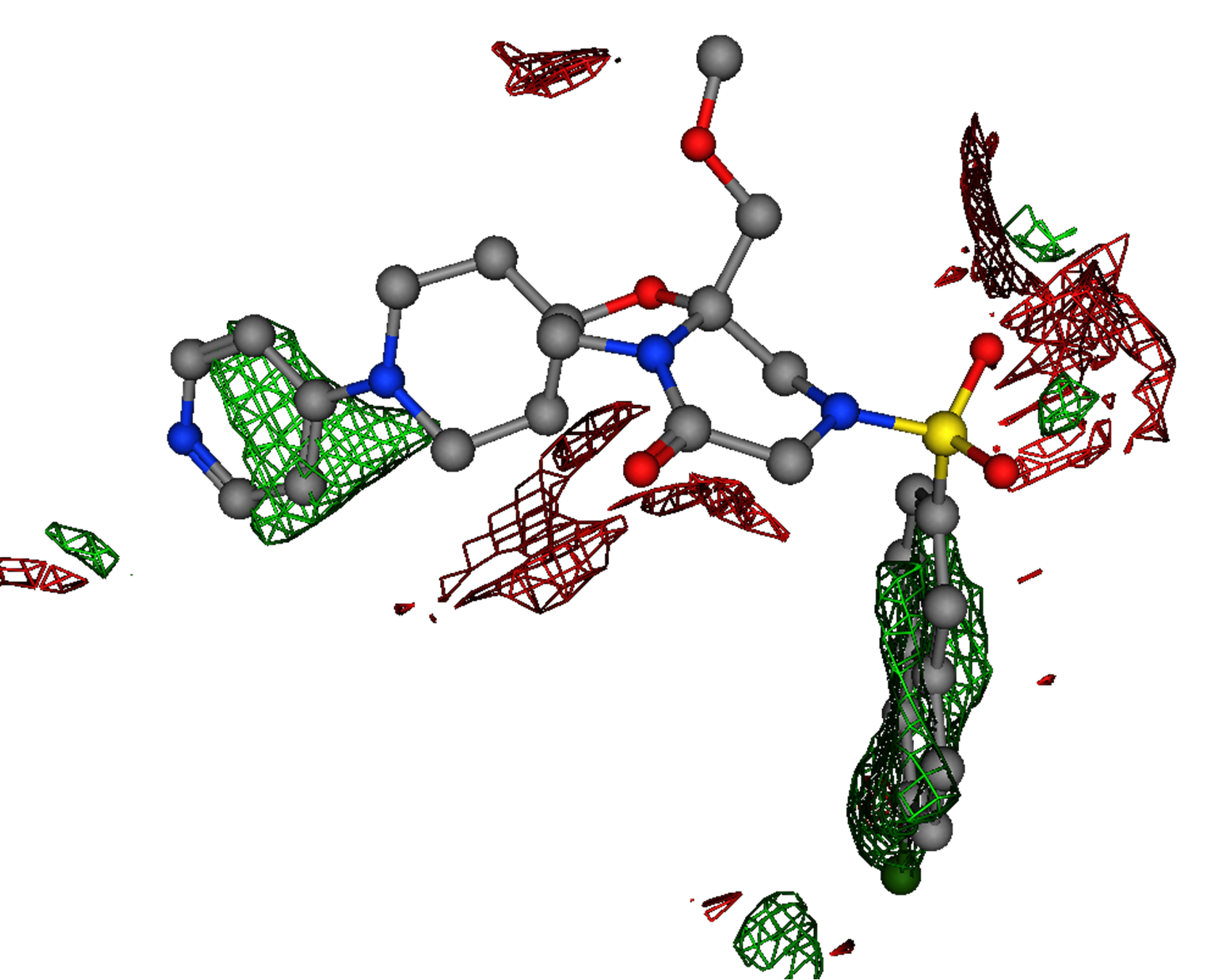
Figure 6.
The electrostatic isocontours of the Factor Xa receptor produced by Electrostatic Maps with the M55532 crystal ligand.
Figure 6 shows the electrostatic isocontours, produced by the Electrostatic
Maps application, superimposed onto the crystal ligand (M55532).
The receptor structure and surface are hidden to illustrate the
correspondence between the prediction and the ligand features.
The strong hydrophobic region overlays with the ligand 4-amino pyridine group
in the D pocket. Notwithstanding the carboxylate near the S1 pocket (Asp189),
there is a large hydrophobic region that overlays well with ligand napthyl
fragment. There are no other strongly hydrophobic regions in the active site.
In addition, the map shows a preference for a negative feature
(red) which overlays with the carbonyl oxygen atom on the ligand.
The electrostatic analysis shows that the strongly interacting groups
are the 4-amino pyridine group in the D pocket and the chloro-naphthyl group
in the S1 pocket. The remainder of M55532 (in the P pocket) can be considered
a “linker” and a good candidate for scaffold replacement.
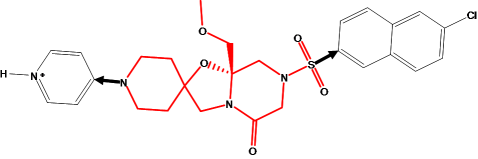
Figure 7.
The M55532 ligand with scaffold drawn with red bonds and R-groups drawn in
black.
The bonds that link the (red) scaffold to the 4-amino pyridine and chloro-napthyl
groups are marked with arrows. A new scaffold should have a conformation
that would, upon substitution, preserve the orientation of the non-scaffold
groups. In particular, the bonds marked with arrows should overlay closely
with the corresponding bonds of a new substituted scaffold.
A Link feature is placed on each scaffold atom that is bonded
to a group to preserve, and a
Link2 feature is placed on each substituent atom bonded to the
existing scaffold. This creates a 4-point pharmacophore query.
The four point query (with 0.3 Å radii for the Link and Link2
features) was used to search a database containing potential
linkers. The database contained the conformations of over 21,372 linkers and
scaffold fragments, which were prepared from more than 40 commercial catalogs
by removing R-groups and other “decorations” based on chemical patterns.
The search returned 8,489 conformations from 1,553 distinct molecules and are
shown overlayed with M55532 in Figure 8.
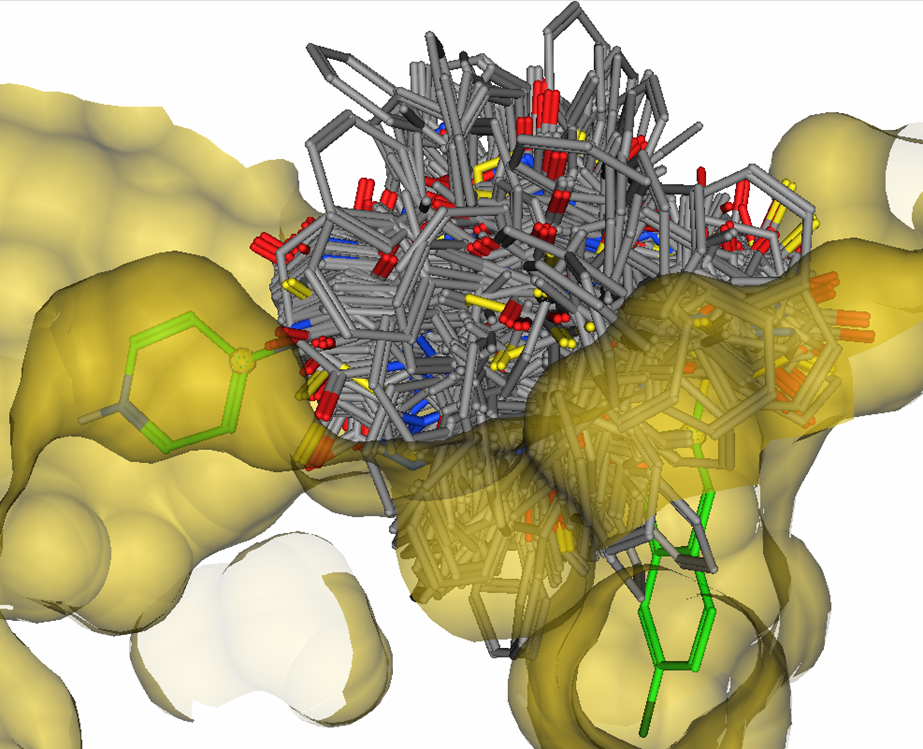
Figure 8.
1,553 candidate linkers overlayed with M55532 in the active site of
Factor Xa, many of which penetrate the van der Waals surface of the
receptor.
In the linker hits, both bond lengths and bond angles are satisfied
for substitution of both the 4-amino pyridine group and the napthalene
group. However, a large number of potential linkers have van der Waals clashes
with the atoms of the receptor or the atoms of the R-groups.
This highlights a key limitation of pure CAVEAT style pharmacophore
queries in which geometric constraints alone are defined and steric
clashes are not considered — a large number of false positives would
be produced.
To avoid such clashes, the pharmacophore query should be augmented with
excluded volume constraints on the R-groups as well as around the binding
site (within 2.2 Å) for modeling receptor shape.
In addition, more traditional features such as hydrogen bond donors
and acceptors can be added to the query. In the case of Factor Xa,
the M55532 ligand had a hydrogen bond to Gly216 and the electrostatic maps
show a strong preference for a negative feature at that location.
The augmented query consists of:
- four link features (Link&Link2 to preserve two bonds); and
- a 1.0 Å acceptor pharmacophore feature (Acc); and
- excluded volumes on the R-groups; and
- a union of excluded volume constraints for receptor shape.
This query results in fewer hits as shown in Figure 9.
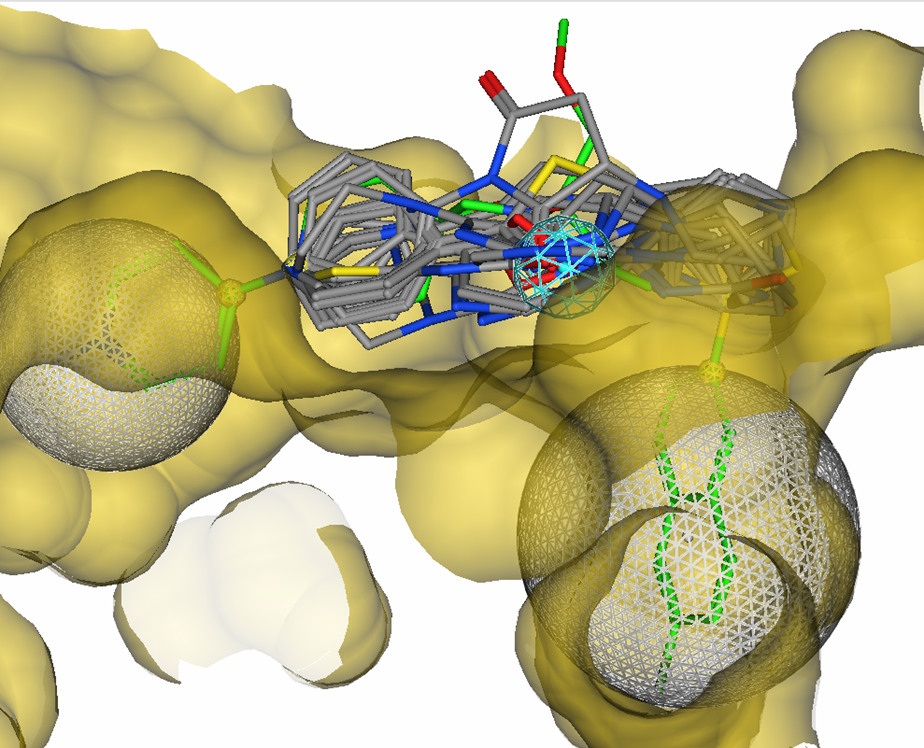
Figure 9.
16 candidate linkers overlayed with M55532 in the active site of
Factor Xa. Excluded volumes and one acceptor feature were used
in a 5-point pharmacophore query.
Table 1 shows some of the returned potential scaffolds. Not
all of the candidate scaffolds connect a nitrogen to the pyridine ring.
This is a problem since the scaffold nitrogen is required to form the
4-amino pyridine group to retain the key basic feature for binding in
the D pocket.
Table 1: Structures of potential linker scaffolds between the napthalene
and pyridil R-groups in M55532
Fortunately, the MOE pharmacophore tools allow for SMARTS patterns in the query.
In this case, a boolean expression is added to ensure that the basic nitrogen is
part of the scaffold. The Link query feature of the scaffold is
replaced with Link & "N". The resulting
query produces two distinct candidate linkers shown below.
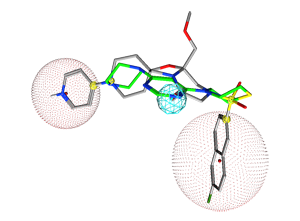
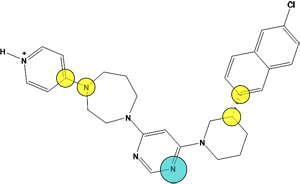
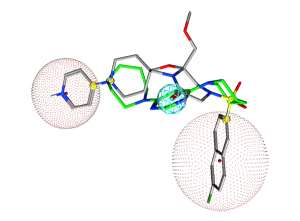
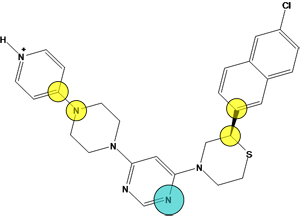
Figure 10.
The pharmacophore query with two cadidate replacement scaffold. The query
consists of a) exit vectors defined by (Link,Link2) pairs drawn in yellow (0.3 Å). The Link feature on the scaffold atom connecting to the pyridyl ring is further specified with Link & "N" to preserve the nitrogen atom, b) an acceptor feature (Acc) drawn in cyan, c) two excluded volumes drawn in red to avoid steric clashes with the R-groups (2.6 Å and 3.6 Å), and d) an excluded volume around the receptor (2.2 Å). In each
case, the native ligand carbons are colored in gray and a candidate replace
scaffold carbons are colored green.
Both candidate linkers preserve the substituent bond vectors, do not clash
with the receptor and contain a nitrogen atom satisfying the requirement for
a hydrogen bond acceptor with Gly216. In addition, the 4-amino pyridine
group is maintained in both scaffolds required for the creation of a salt bridge with
Glu97.
We have shown how MOE's pharmacophore tools are used to perform scaffold
replacement experiments. In similar fashion to CAVEAT, an exit vector
is defined using pairs of special “Link” pharmacophore features.
A pair of Link features on a scaffold atom and a connected atom from the
R-group are defined for each bond that needs to be preserved.
Choosing R-groups is done by identifying key interactions between the native
ligand and the receptor.
Active site analysis tools such as Ligand Interactions, Contact Statistics,
and Electrostatic Maps can also act as a guide in choosing appropriate R-groups.
MOE's pharmacophore tools show several advantages to traditional computational
scaffold replacement techniques. Unlike the pure CAVEAT methodology alone,
MOE's Link features can be used in conjunction with other query features,
as well as volumes to create more sophisticated queries that preserve important
scaffold interactions or avoid van der Waals clashes with the receptor.
Ad hoc SMARTS expressions can be incorporated to enforce specific
chemical group requirements. In addition, no special preparation is
required for the linker database — any 3D conformation database can be
searched.
The combination of active site analysis tools and pharmacohpore-based
scaffold replacement methods means that scaffold replacement can be
routinely performed in structure-based design projects.
| [Clark 2007]
|
Clark, A.M; Labute, P.
2D Depiction of Protein-Ligand Complexes
J. Chem. Inf. Model. 47 (2007 1933-1944.
|
| [Davie 1991]
|
Davie, E.W; Fujikawa, K.; Kisiel, W.;
The Coagulation cascade: initiation, maintenance, and regulation.
Biochemistry 30 (1991) 10363-1-370.
|
| [Halgren 1996]
|
Halgren, T.A.;
The Merck Force Field;
J. Comp. Chem. 17 (1996) 490-641.
|
| [Kastenholz 2000]
|
Kastenholz, M.A.; Pastor, M.; Cruciani, G.; Haaksma, E.E.J.; Fox, T.;
GRID/CPCA: A New Computational Tool to Design Selective Ligands.
J. Med. Chem. 43 (2000) 3033-3044.
|
| [Lauri 1994]
|
Lauri, G., Bartlett, P. A.;
CAVEAT: A Program to Facilitate the Design of Organic Molecules;
J. Comp. Aided Mol. Des. 8 (1994) 51-66.
|
| [Lewell 1998]
|
Lewell, X.Q., Judd, D.B., Watson, S.P., Hann, M.M.;
RECAP — Retrosynthetic Combinatorial Analysis Procedure: A Powerful New
Technique for Identifying Privileged Molecular Fragments with Useful
Applications in Combinatorial Chemistry;
J. Chem. Inf. Comput. Sci. 38 (1998) 511-522.
|
| [Maass 2007]
|
Maass, P.; Schulz-Gasch, T.; Stahl, M.; Rarey, M.;
Recore: A Fast and Versatile Method for Scaffold Hopping Based on Small
Molecule Crystal Structure Conformations;
J. Chem. Inf. Model. 27 (2007) 390-399.
|
| [Takano 2007]
|
Takano Y.; Koizumi M.; Takarada R.; Takimoto Kamimura M.; Czerminski R.; Koike
T.;
Computer-aided design of a factor Xa inhibitor by using MCSS functionality
maps and a CAVEAT linker search;
Journal of Molecular Graphics and Modelling 22 (2003)
105-114.
|
| [Stubbs 1995]
|
Stubbs, M.T.; Huber, R.; Bode, W.;
Crystal Structure of Factor Xa Specific Inhibitors in Complex with Trypsin:
Structural Grounds for Inhibition of Factor Xa and Selectivity Against Thrombin.
FEBS Lett. 375 (1995) 103-107.
|
|